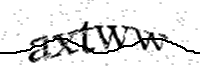Validation request
User validation required to continue..
Please type the text you see in the image into the text box and submit